Review on electrochemical simulations published in Chem. Rev.
R. Sundararaman, D. Vigil-Fowler and K. Schwarz, “Improving the Accuracy of Atomistic Simulations of the Electrochemical Interface”, Chem. Rev. 122, 10651 (2022)
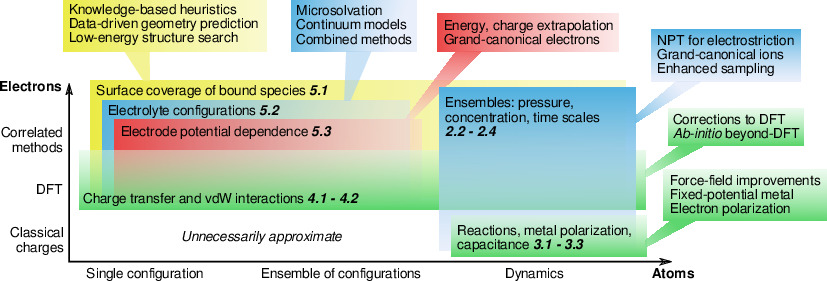
Atomistic simulation of the electrochemical double layer is an ambitious undertaking, requiring quantum mechanical description of electrons, phase space sampling of liquid electrolytes, and equilibration of electrolytes over nanosecond timescales. All models of electrochemistry make different trade-offs in the approximation of electrons and atomic configurations, from the extremes of classical molecular dynamics of a complete interface with point-charge atoms to correlated electronic structure methods of a single electrode configuration with no dynamics or electrolyte. Here, we review the spectrum of simulation techniques suitable for electrochemistry, focusing on the key approximations and accuracy considerations for each technique. We discuss promising approaches, such as enhanced sampling techniques for atomic configurations and computationally-efficient beyond density functional theory (DFT) electronic methods, that will push electrochemical simulations beyond the present frontier.