Improved method for potentials in DFT published in J. Chem. Phys.
R. Sundararaman and Y. Ping, “First-principles electrostatic potentials for reliable alignment at interfaces and defects”, J. Chem. Phys. 146, 104109 (2017) (Preprint: arXiv:1612.01671)
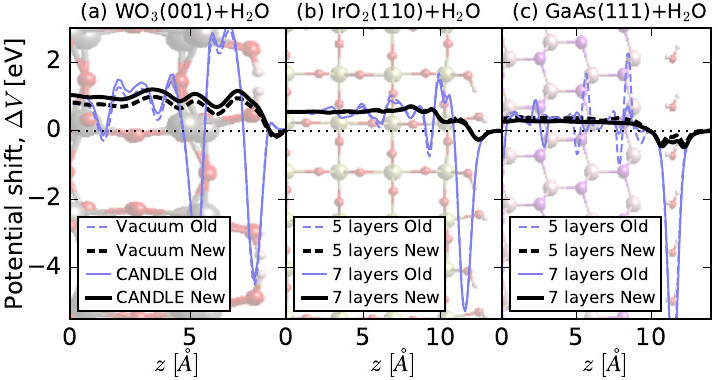
Our team at UC Santa Cruz and RPI developed a new strategy to calculate electrostatic potentials in DFT calculations, which eliminates strong oscillations at the atomic scale that plague the conventional approach. This allows us to calculate potential differences at interfaces and near defects more accurately than before, substantially simplifying the calculation of band alignment across interfaces and formation energies of charged defects. Combined with our solvation methodology, this makes it now possible for us to investigate charged defects at solid-liquid interfaces.