Non-equilibrium carrier dynamics
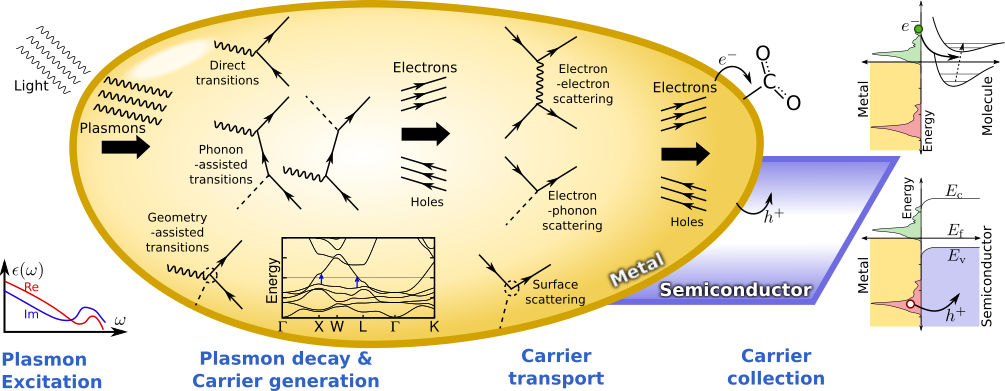
Conventional solar energy and electronic devices typically work with electrons and holes in semiconductors that are close to equilibrium. In nanoscale systems, it is possible to access these charge carriers while they are still far from equilibrium, eg. hot carriers excited by photons absorbed in plasmonic nanostructures. Avoiding equilibration could provide a pathway for higher-efficiency light capture for solar energy conversion, higher sensitivity for photodetection and lower loss in electronic devices.
We develop methods based on density-functional theory to calculate the electronic structure, electron-phonon interactions and electron-light interactions of materials, and thereby predict the non-equilibrium dynamics and transport of excited carriers in nano-scale systems. For example, we now predict the femtosecond dynamics and optical response of such carriers in quantitative agreement with pump-probe spectroscopy experiments. Armed with these methods, we will target the inverse problem: which combinations of materials and nanostructures generate the optimum carrier distributions for a given application?
Atomic-scale liquid response
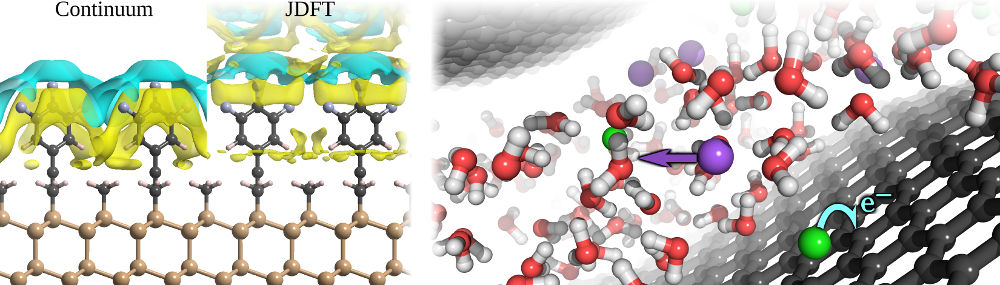
Solid-liquid interfaces are integral to biological systems, catalysis and energy conversion & storage systems including batteries, fuel cells & supercapacitors. The complex interactions between electronic properties of the solid, atomic-scale response of the liquid at the interface and longer-range properties of the liquid make such systems challenging for experimental characterization as well as theoretical modelling. Joint density-functional theory (JDFT) combines electronic structure methods with statistical theories of liquids to efficiently and accurately predict properties of solid-liquid interfaces, which are notoriously difficult to simulate using conventional methods based on molecular dynamics.
JDFT-based methods can predict the alignment of electronic bands across solid-liquid interfaces, which is a critical property for efficient photoelectrochemical energy conversion. Using these methods, we can design surface terminations of photoelectrodes to optimize band alignments, independent of the light capture and carrier transport properties of the electrode, giving us additional knobs to improve the overall efficiency of such energy conversion. Further, we can examine the effect of nano-scale confinement of the liquid on chemical reaction and electron transfer rates, potentially providing yet another microscopic control on the properties of these interfaces. Finally, time-dependent extensions of JDFT will make it possible to explore the non-equilibrium charge transport of ions in the liquid, which is of fundamental importance to chemical dynamics in general, and the performance of batteries and supercapacitors in particular.
Electronic-structure methods & software
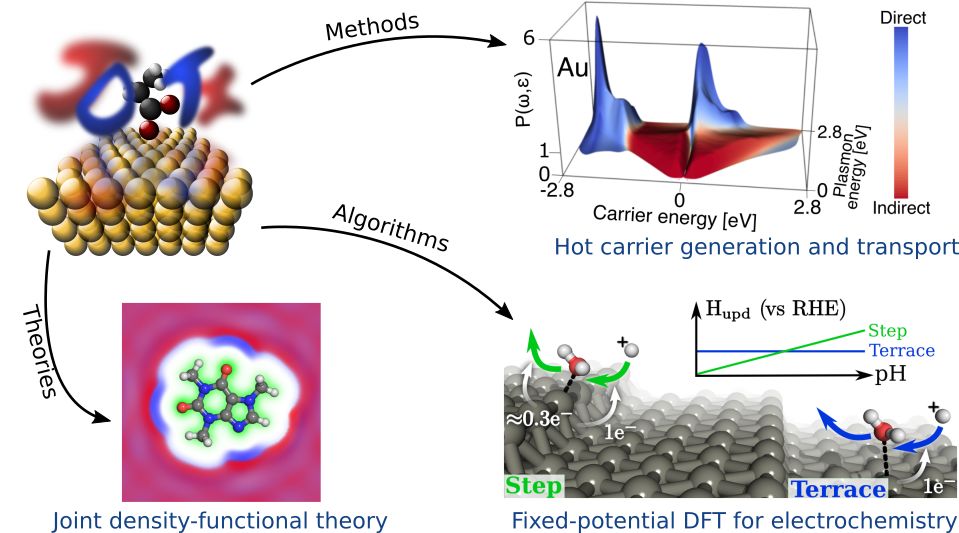
At the heart of predictive computation for nanoscale systems lie new theoretical models, algorithms and software. We lead the development of the open-source DFT software, JDFTx, that enables rapid development of new models via an object-oriented framework within which a single implementation performs well on a variety of high-performance computing hardware. For example, we have developed several accurate theoretical models for liquids, electron-liquid interactions and electronic structure.
JDFTx was initially developed for joint-density functional theory (JDFT), and has supported the development of a heirarchy of methods to describe electronic structure of systems with liquids. Within JDFTx, we develop new algorithms eg. to treat electrochemical systems at fixed potential instead of at fixed electron number in order to match the experimental conditions. More generally, JDFTx is now the ideal platform to develop methods that combine quantum-mechanical DFT calculations with longer-range theories, including for electronic and liquid structure, electron-phonon and electron-photon interactions, and for non-equilibrium carrier dynamics.