Universal machine-learned 1D DFT functionals published in J. Chem. Phys.
M. M. Kelley, J. Quinton, K. Fazel, N. Karimitari, C. Sutton and R. Sundararaman, “Bridging electronic and classical density-functional theory using universal machine-learned functional approximations”, J. Chem. Phys. 161, 144101 (2024)
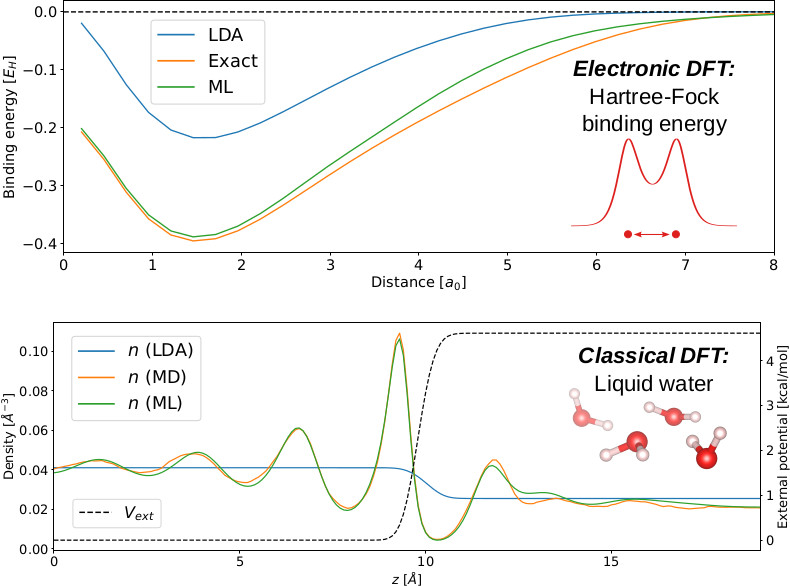
The accuracy of density-functional theory (DFT) calculations is ultimately determined by the quality of the underlying approximate functionals, namely the exchange-correlation functional in electronic DFT and the excess functional in the classical DFT formalism of fluids. For both electrons and fluids, the exact functional is highly nonlocal, yet most calculations employ approximate functionals that are semi-local or nonlocal in a limited weighted-density form. Machine-learned (ML) nonlocal density-functional approximations show promise in advancing applications of both electronic and classical DFTs, but so far these two distinct research areas have implemented disparate approaches with limited generality. Here, we formulate a universal ML framework and training protocol to learn nonlocal functionals that combine features of equivariant convolutional neural networks and the weighted-density approximation. We prototype this new approach for several 1D and quasi-1D problems and demonstrate that functionals with exactly the same hyperparameters achieve excellent accuracy for a diverse set of systems, including the hard-rod fluid, the inhomogeneous Ising model, the exact exchange energy of electrons, the electron kinetic energy for orbital-free DFT, as well as for liquid water with 1D inhomogeneities. These results lay the foundation for a universal ML approach to approximate exact 3D functionals spanning electronic and classical DFTs.