First-principles NaCl melting simulations featured in J. Chem. Phys.
T. Shah, K. Fazel, J. Lian, L. Huang, Y. Shi and R. Sundararaman, “First-principles molten salt phase diagrams through thermodynamic integration”, J. Chem. Phys. 159, 124502 (2023)
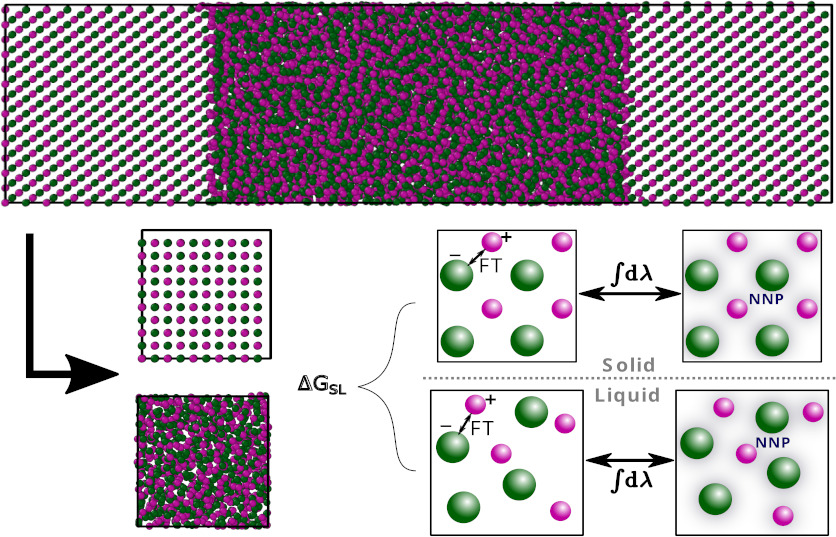
Precise prediction of phase diagrams in molecular dynamics simulations is challenging due to the simultaneous need for long time and large length scales and accurate interatomic potentials. We show that thermodynamic integration from low-cost force fields to neural network potentials trained using density-functional theory (DFT) enables rapid first-principles prediction of the solid–liquid phase boundary in the model salt NaCl. We use this technique to compare the accuracy of several DFT exchange–correlation functionals for predicting the NaCl phase boundary and find that the inclusion of dispersion interactions is critical to obtain good agreement with experiment. Importantly, our approach introduces a method to predict solid–liquid phase boundaries for any material at an ab initio level of accuracy, with the majority of the computational cost at the level of classical potentials.
This article was selected to be a feature article in J. Chem. Phys..